
Our group is entirely computational. We collaborate with experimental groups to generate and analyze genetic variation data. Additionally, we make extensive use of existing publicly-available datasets.
Our work consists of both developing new methods and well as applying existing methods to new datasets. Though our work is computational, our focus lies in addressing interesting biological questions where new, careful analyses can yield novel and interesting insights.
While much of Kirk’s previous work has involved studying genetic variation in humans, we are also currently studying organisms across the tree of life. Recent projects have focused on sea otters, dogs, wolves, foxes, and plants.
Current work in the lab is focused in three main areas:
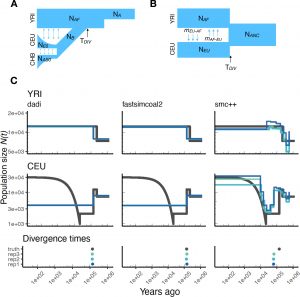 We develop and implement statistical approaches to estimate demographic parameters from genetic variation data. Current projects are focused on collaborating with empirical researchers to apply population genetic tools to infer demography of a variety of species including California Oak trees, sea otters, the endangered vaquita, and the finn whale. An additional area of research focuses on assessing the performance of population genetic methods to infer demographic history in complex models. We are part of the PopSim Consortium which is creating a library implementing previously published demographic models in msPrime. This project should enable more robust benchmarking of inference methods.
We develop and implement statistical approaches to estimate demographic parameters from genetic variation data. Current projects are focused on collaborating with empirical researchers to apply population genetic tools to infer demography of a variety of species including California Oak trees, sea otters, the endangered vaquita, and the finn whale. An additional area of research focuses on assessing the performance of population genetic methods to infer demographic history in complex models. We are part of the PopSim Consortium which is creating a library implementing previously published demographic models in msPrime. This project should enable more robust benchmarking of inference methods.
Inference of natural selection
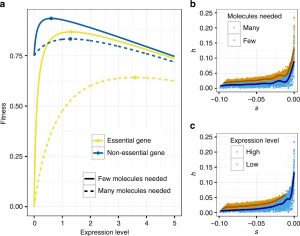 Negative natural selection is the process where deleterious mutations are removed from the population. We use population genetic models and empirical data to study how this process works. We are specifically interested in how population history influences negative selection and how negatively selected sites affect linked neutral variation (background selection), patterns of admixture, and sequence constraint across species. Current work also involves learning about the distribution of fitness effects (DFE). To this end, we have developed tools to infer the DFE and have compared the DFE across species. Further, we are now developing approaches to infer the DFE that leverage more complex patterns of the data to better infer dominance of deleterious mutations and how selection may have changed over time. Lastly, we develop conceptual and theoretical models for the differences in the DFE of deleterious mutations across species, the proportion of beneficial mutations across species, and the reasons for the existence of dominance. The figure to the left shows our conceptual model for dominance that can explain many features of the data, such as the finding that more deleterious mutations tend to be more recessive and that highly expressed genes tend to be more additive that more lowly expressed genes.
Negative natural selection is the process where deleterious mutations are removed from the population. We use population genetic models and empirical data to study how this process works. We are specifically interested in how population history influences negative selection and how negatively selected sites affect linked neutral variation (background selection), patterns of admixture, and sequence constraint across species. Current work also involves learning about the distribution of fitness effects (DFE). To this end, we have developed tools to infer the DFE and have compared the DFE across species. Further, we are now developing approaches to infer the DFE that leverage more complex patterns of the data to better infer dominance of deleterious mutations and how selection may have changed over time. Lastly, we develop conceptual and theoretical models for the differences in the DFE of deleterious mutations across species, the proportion of beneficial mutations across species, and the reasons for the existence of dominance. The figure to the left shows our conceptual model for dominance that can explain many features of the data, such as the finding that more deleterious mutations tend to be more recessive and that highly expressed genes tend to be more additive that more lowly expressed genes.
Understanding the genetic basis of complex traits
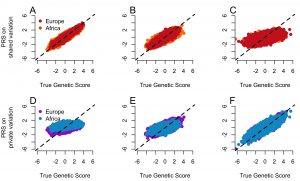 Common diseases are caused, in part, by many genetic factors. Efforts are underway to find the variants responsible and use these variants in risk prediction. Our work takes advantage of population genetics to better inform such mapping studies. In particular, we have examined how negative natural selection and population history influence the architecture (e.g. the number of causal variants, their frequencies, and their effect sizes) of complex traits. For example, we recently found that private variants (e.g. variants that are only polymorphic in one population) may account for at least 20% of the heritability of complex traits. The figure to the left shows that such a trait architecture will lead to poor transferability of risk scores across populations. This is one possible reason why risk scores do not transfer well across populations. Ongoing projects are aimed at quantifying the extent of selection on complex traits and improving genetic risk prediction in diverse populations.
Common diseases are caused, in part, by many genetic factors. Efforts are underway to find the variants responsible and use these variants in risk prediction. Our work takes advantage of population genetics to better inform such mapping studies. In particular, we have examined how negative natural selection and population history influence the architecture (e.g. the number of causal variants, their frequencies, and their effect sizes) of complex traits. For example, we recently found that private variants (e.g. variants that are only polymorphic in one population) may account for at least 20% of the heritability of complex traits. The figure to the left shows that such a trait architecture will lead to poor transferability of risk scores across populations. This is one possible reason why risk scores do not transfer well across populations. Ongoing projects are aimed at quantifying the extent of selection on complex traits and improving genetic risk prediction in diverse populations.